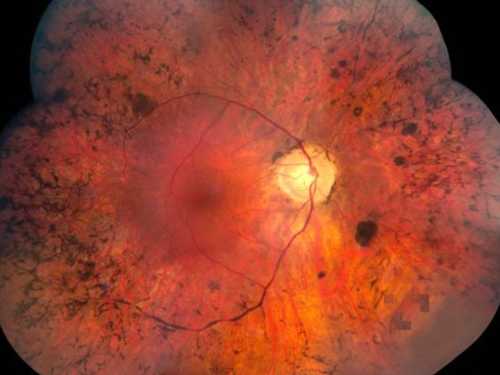
La Retinite pigmentosa (RP) è una distrofia retinica ereditaria, causata dalla perdita dei fotorecettori e caratterizzata da depositi retinici di pigmento visibili all’esame del fondo dell’occhio.
Si stima che 1,39 milioni di individui nel mondo ne siano affetti. La malattia è ereditaria e il suo decorso naturale porta alla progressiva perdita della vista. E’ classificata tra le malattie non curabili ed è la terza causa di handicap visivo, la prima in pazienti al di sotto dei 60 anni di età.
La forma più comune è la distrofia tipo bastoncelli-coni, che insorge con cecità notturna, seguita dalla perdita progressiva della vista diurna, del campo visivo periferico, che può portare a cecità dopo diverse decadi.
Generalmente, la RP non è sindromica anche se sono note diverse forme associate a sindromi, la più frequente delle quali è la sindrome di Usher (associata a sordità congenita profonda). Sono stati identificati circa 50 geni/loci responsabili della RP non sindromica (forme autosomiche dominanti, autosomiche recessive, legate all’X e digeniche).
DIAGNOSI
- presenza di cecità notturna
- fotofobia e abbagliamento eccessivo
- limitazioni del campo visivo
- lesioni tipiche nel fondo dell’occhio
- ERG ipovoltato
La diagnosi molecolare è possibile per alcuni geni, ma, di regola, non viene eseguita a causa dell’estrema eterogeneità genetica della malattia. La consulenza genetica è sempre indicata.
TERAPIA
Al momento, non è disponibile una terapia in grado di arrestare la progressione della malattia o di restituire la vista; pertanto, la prognosi è infausta. L’approccio terapeutico permette di rallentare il processo degenerativo e consiste nella protezione dalla luce solare e nella terapia vitaminica, nel trattamento delle complicanze (cataratta e edema maculare) e nell’aiutare i pazienti a fare fronte ai problemi sociali e psicologici correlati alla cecità.
RICERCA
La ricerca scientifica è molto attiva e stanno emergendo nuove strategie terapeutiche stanno emergendo nuove strategie terapeutiche che spaziano dalla terapia genica alla neuroprotezione alle protesi retiniche. In questo constesto si è appena conclusa in Giappone la fase 3 dello studio clinico sull’efficacia e la sicurezza dell’Unoprostone (UF-021) in soluzione oftalmica per il trattamento della retinite pigmentosa.
Lo studio si è svolto nell’arco di due anni consecutivi su 180 pazienti trattati in 38 centri in Giappone. L’endpoint principale dello studio era la modificazione dei valori di sensibilità retinica media su quattro punti centrali , rilevata attraverso Humphrey Field Analyzer (HFA). Rispetto al gruppo trattato con placebo, la differenza non è risultata statisticamente significativa.
E’ stato tuttavia rilevato un miglioramento statisticamente significativo della sensibilità retinica e dell’acuità visiva rispetto al giorno 0 dello studio nel gruppo trattato con UF-021, a fronte di una progressiva riduzione del campo visivo (Goldmann) nel gruppo di controllo.
Si è registrato inoltre un incremento statisticamente significativo del punteggio totale del VFQ-25 nel gruppo UF-021. I prostoni sono una classe di acidi grassi con effetto simile a quello delle prostaglandine, ma privi degli effetti collaterali delle stesse.
Usati come farmaci antiglaucomatosi per le loro proprietà ipotensive, si sono dimostrati in grado di esplicare un’azione protettiva sui nervi ottici in vitro e di migliorare la microcircolazione oculare nel glaucoma a pressione normale.